Applications of Coarse-Grained Models for DNA
Molecular modeling has played a vital role in developing the understanding of the structure and dynamics of DNA. Traditional all-atom representations of DNA have been very successful, but their complexity limits the time and length scales that can be probed, constraining the range of problems that can be considered. Therefore, we have pioneered the development and application of coarse-grained molecular models of DNA, where each nucleotide is replaced by just two or three force sites. These simple models enable us to examine systems that are inaccessible by traditional atomic descriptions.
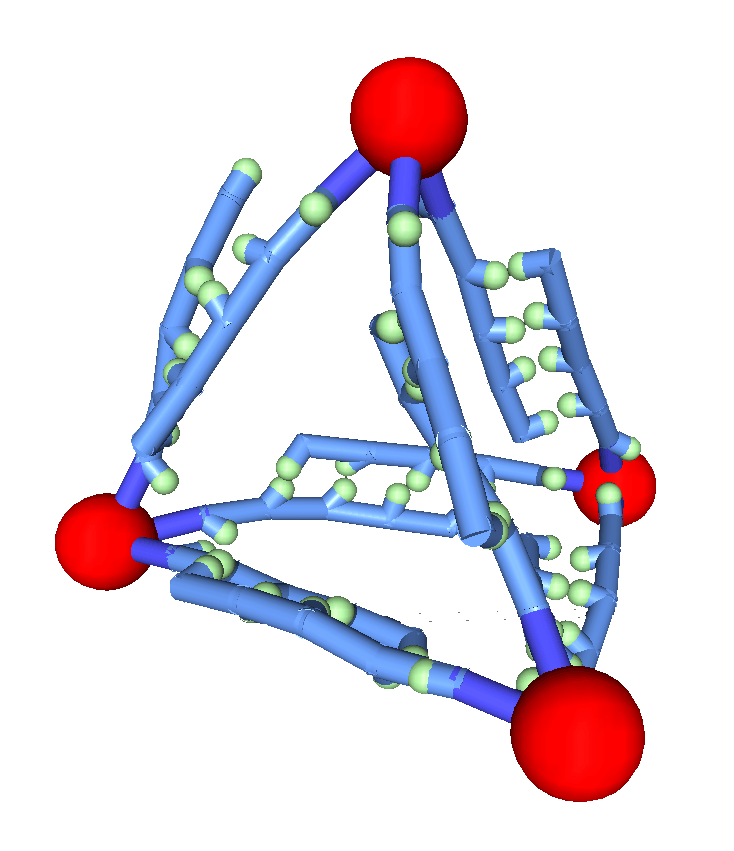
Coarse-grained representations of DNA are ideal to study the intersection of biophysics and nanotechnology, where DNA is used to create synthetic nanostructures and nanomaterials. For example, by linking multiple single-stranded DNA to a core nanoparticle, we create a designer “atom” with controllable binding energy, bond length, and valence, which can then serve as a node for complex, higher-order structures. Using our coarse-grained models, we have shown how these “atoms” spontaneously self-assemble into nanomaterials with phase behavior that is unique in comparison to naturally occurring matter. Our studies also examine the theory needed to comprehend the thermodynamics and dynamics of these new materials. These synthetic systems can also be informative about naturally occurring systems. For example, nanoparticles with four tethered single strands of DNA have offered a mechanism for the emergence of liquid-liquid transitions in important natural systems like water and silica.

We are also using coarse-grained models to probe problems of direct biological importance. For example, in collaboration with experimental studies, we examine how coarse-grained models can be use to predict the active regions in 4-way DNA junctions, and quantitatively predict both melting temperatures and the underlying molecular mechanism for melting. Looking to the future, we aim to examine how these coarse-grained representations can be combined with description of proteins to tackle very large-scale problems, such as the formation of chromatin.
Polymers, Nanoparticle Composites, and Ultra-Thin Films
 The addition of organic or inorganic material to polymers has long been exploited to change material properties. With the emergence of custom designed nanoparticles, it has been shown that dramatic changes in material properties with only a fraction of added nanoparticles. To achieve these improvements, it is critical to control the clustering behavior of the added nanoparticles.
The addition of organic or inorganic material to polymers has long been exploited to change material properties. With the emergence of custom designed nanoparticles, it has been shown that dramatic changes in material properties with only a fraction of added nanoparticles. To achieve these improvements, it is critical to control the clustering behavior of the added nanoparticles.
Our recent work has shown that the clustering of nanoparticles appears to be described by the same physics controlling equilibrium polymerization and thermoreversible gelation. Unfortunately, the minimal criteria for a reversible gelation and the detailed physics of gelation are not nearly as well understood as simple phase transitions, which are well described by statistical physics.
We are studying models that reveal insights into the basic phenomena of gelation, as well as models that elucidate practical connections between interfacial nanoparticle-polymer behavior and bulk properties. We are also using the insights from these systems to improve the modeling of nanocomposite materials.
Cooperative Dynamics of Lipid Membranes and Monolayers
Lipid monolayers, membranes, and droplets play an integral role in dynamic cellular processes, and improper function of these structures is related to numerous health issues, including impaired lung function, cardiovascular disease, Alzheimer’s disease, and cancer. Yet the dynamic characteristics of these ubiquitous structures remain poorly understood. It is widely appreciated that dynamic heterogeneity of lipid structures is essential to their function, qualitatively captured by the contentious “lipid-raft” concept, the primary paradigm for lipid structure-function relation. Yet the definition and quantification of the dynamically heterogeneous structures remains an ongoing challenge.
Our research on lipid membranes draws on methods from condensed matter physics to provide a quantitative framework to describe dynamical lipid structures. Due to the similarity of physical interactions between lipid structures and other condensed materials, lipid layers apparently display similar dynamic heterogeneity. Indeed, single-component membranes can exhibit dynamical structures with the length and time scales expected of more complex rafts, strongly suggesting that the underlying and general dynamic heterogeneity of membranes plays an important role in the organization of complex raft structures that facilitate membrane function. A new framework for lipid dynamic association would impact our understanding of dynamic processes, such as protein insertion, self-assembly, endocytosis and exocytosis.
Supercooled and Glassy Water
 While water may be ubiquitous, it is still relatively poorly understood in comparison to “simple” liquids. Not surprisingly, this is largely due to hydrogen bonding, which gives rise to the well-known fact that ice floats in liquid water. These odd behaviors become even more pronounced in the supercooled region of liquid water — that is, when the liquid is cooled below its equilibrium freezing point.
While water may be ubiquitous, it is still relatively poorly understood in comparison to “simple” liquids. Not surprisingly, this is largely due to hydrogen bonding, which gives rise to the well-known fact that ice floats in liquid water. These odd behaviors become even more pronounced in the supercooled region of liquid water — that is, when the liquid is cooled below its equilibrium freezing point.
Our research focuses on the behavior of water in this supercooled region, focusing on the interplay between thermodynamics and dynamics that is not apparent at higher temperatures. More specifically, simulations suggest that there is a low temperature phase transition between two distinct liquid states. As a result, it appears that the dynamic properties in the vicinity of that transition differ significantly from other liquids. In addition, we also study the behavior of water confined to nanoscale regions. The properties of this confined water can differ greatly from the behavior of ordinary water, which has important implications in nanofluidic and biological systems.
Glass Transition
The process by which an amorphous liquid becomes an amorphous solid (as opposed to a crystalline solid) has been a subject of study for millennia. While the understanding of the gradual slowing of liquid dynamics has greatly improved, there remain many open questions.
Computational studies are particularly useful to identify the molecular level processes that are involved in the relaxation of liquids near their glass transition. Computational models have played a major role in developing the “energy landscape” approach of liquid dynamics, the phenomena of dynamic heterogeneity, and in showing the connection between local structure of the liquid and its dynamics. Our research aims at developing each of these inter-related approaches.